 如何同時使用Nucleus與TensorFlow解決基因組學領域的機器學習問題
如何同時使用Nucleus與TensorFlow解決基因組學領域的機器學習問題
簡介
在本文中,我們將 DNA 測序糾錯表述為多級分類問題,并提出兩種深度學習解決方案。第一種方法是在單次讀取中糾錯,而第二種方法(如圖 1 所示)則通過多次讀取來達成共識,以預測正確的 DNA 序列。我們的Colab 筆記教程使用Nucleus和TensorFlow庫實現第二種方法。本文旨在向您展示如何同時使用 Nucleus 與 TensorFlow 解決基因組學領域的機器學習問題。

問題概覽
盡管 DNA 測序日漸快捷和便宜,其過程仍容易出錯。使用Illumina等公司開發的新一代測序 (NGS) 技術處理原始數據時,錯誤率約為 1%。第三代技術,例如Pacific BioSciences(PacBio) 公司開發的技術,正日益普及,其錯誤率約為 15%。測序錯誤可分為替換、插入和缺失,后兩者通常稱為 indel。所有這些錯誤均不利于下游的分析步驟,例如變異檢測和基因組組裝。
如要獲取較高質量的數據集,一個簡單的方法是舍棄可能包含錯誤的數據,丟棄全部讀取內容或去除低質量區域皆可。該方法并非理想之選,因為這會導致最終的數據集會變小。此外,某些序列上下文本來就有較高的錯誤率,進而導致采樣出現偏差。因此,大量研究都側重于開發更成熟的糾錯方法。大多數已開發的方法均可歸類為以下兩組之一:
對單次讀取進行操作的方法,旨在確定正確的讀取序列
對多次讀取進行操作的方法,以共識為基礎,旨在確定正確的基礎 DNA 序列
深度學習概覽
本文中闡述的兩種方法均使用深度神經網絡,學習將輸入映射至輸出的函數。神經網絡由若干層線性與非線性運算構成,而這些運算會依次應用至輸入。神經網絡已成功應用于包括圖像分類和自然語言翻譯在內的多個問題領域。最近,神經網絡也被用于解決基因組學問題,例如蛋白質結構預測和變異檢測。
方法
Nucleus
我們的實現需要Nucleus,這是由 Google Brain 的 Genomics 團隊開發的內容庫,用于處理基因組學數據。Nucleus 使用專門的 reader 對象與 writer 對象,可輕松讀取、寫入和分析常見基因組文件格式(如 BAM、FASTA 和 VCF)中的數據。Nucleus 讓我們能夠:
針對指定基因組區域中的所有變異查詢 VCF 文件
針對映射至指定基因組范圍的所有讀取內容查詢 BAM 文件
針對從指定位置開始的參考序列查詢 FASTA 文件
我們還能使用 Nucleus 將數據寫入TFRecords,這種二進制文件格式由協議緩沖區消息構成,可由 TensorFlow 輕松讀取。讀取 TFRecords 文件后,我們會使用Estimator API訓練和評估卷積神經網絡。
數據
以下是我們在實現過程中所使用的文件列表。所有數據均公開提供,且此 教程包含下載鏈接與說明。
NA12878_sliced.bam — 從 20 號染色體(位置 10,000,000–10,100,000)獲得的 Illumina HiSeq 讀取內容,降采樣至 30x 的覆蓋度
NA12878_sliced.bam.bai — NA12878_sliced.bam 的索引
NA12878_calls.vcf.gz — 瓶中基因組 NA12878 變異的真值集合
NA12878_calls.vcf.gz.tbi — NA12878_calls.vcf.gz 的索引
hs37d5.fa.gz — hs37d5 的參考基因組
hs37d5.fa.gz.fai 和 hs37d5.fa.gz.gzi — hs37d5.fa.gz 的索引文件
注:教程 鏈接
https://colab.research.google.com/github/google/nucleus/blob/master/nucleus/examples/dna_sequencing_error_correction.ipynb
網絡架構
卷積神經網絡通常用于處理計算機視覺任務,但也非常適用于基因組學。每個卷積層都會反復將學習后的過濾器應用于輸入數據。在網絡中早期出現的卷積過濾器會學習識別輸入數據的低級特征(如圖像中的邊緣及色彩梯度),而后期出現的過濾器則會學習識別更復雜的低級特征組合。對于 DNA 序列輸入,低級卷積過濾器會充當 motif 檢測器,這類似于序列標識圖的位置權重矩陣。
在實現過程中,我們使用的標準卷積架構依次由兩個卷積層及三個全連接層組成。我們使用非線性 ReLU 層提升模型的表現能力。當卷積層減少輸入量后,我們會進行最大池化,并會在全連接層充當正則化矩陣后退出此過程。請注意,在得到最終的全連接層后,我們不會加入 softmax 層,因為我們使用的損失函數是在內部應用 softmax。如需了解每層的詳情,請參閱此教程。
注:教程 鏈接
https://colab.research.google.com/github/google/nucleus/blob/master/nucleus/examples/dna_sequencing_error_correction.ipynb
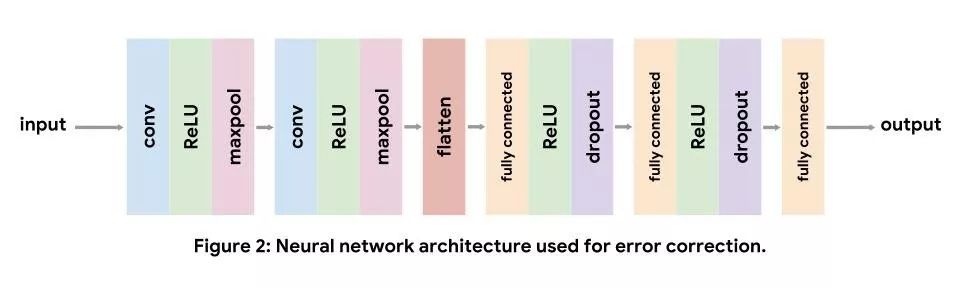
方法 1:單次讀取的糾錯
為了糾正序列讀取中的錯誤,我們使用深度學習來訓練神經網絡,以解決一個較為普遍的問題:填充 DNA 序列中缺失的堿基。此方法旨在開發一種可理解 DNA 序列語法的模型。若僅靠真實序列的語法,我們可能無法獲取充足的信息來開發可用于生產環境的解決方案。盡管如此,這依然是一個簡單明了的示例應用。
出于指導目的,我們通過以下方法簡化此問題:
僅考慮存在替換錯誤的區域,并忽略 indel 錯誤
僅考慮未存在已知變異的區域
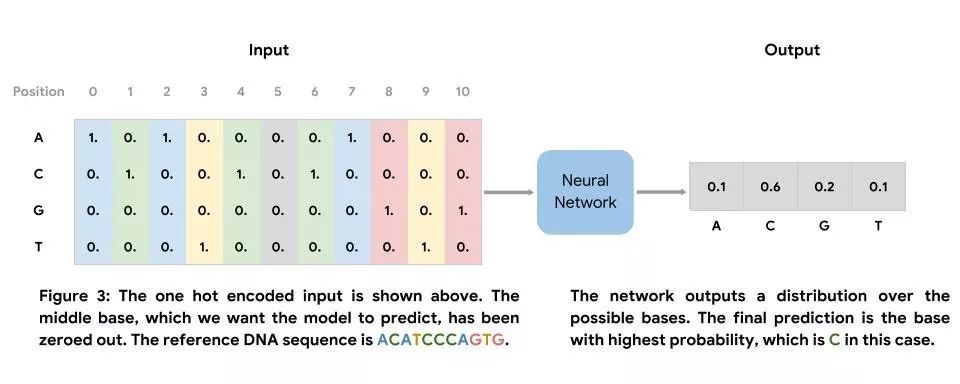
我們可以在參考基因組的區域中訓練該神經網絡。此網絡的輸入是定長的 DNA 序列,其核心是我們希望預測的堿基。此網絡的輸出是可能出現的堿基分布,且最終預測結果為可能性最高的堿基。我們使用在參考基因組中觀測到的堿基產生標簽集。由于我們僅使用映射至未存在已知真值變異之區域的讀取內容,因此可以將參考基因組中存在的堿基明確標記為標簽。
我們將參考基因組分割成非重疊的定長片段,以產生輸入序列。在訓練、評估和測試時,我們將參考序列中的一個堿基置零,以模擬缺失的堿基,如圖 3 所示(位置 5)。除了使用參考基因組來模擬缺失的數據之外,我們還可將此類模型應用于序列讀取的數據,特別是質量評分低于閾值的堿基。
方法 2:基于共識的糾錯
糾錯的最終目的是確定基礎 DNA 序列,而非為了糾正單次讀取的錯誤。在本部分,我們通過匯總序列堆疊來使用多次讀取達成的共識。如此一來,無需糾正單次讀取的中間步驟即可直接確定 DNA 序列。有關序列堆疊的示例如下方圖 4 所示。請注意,下圖僅展示了此窗口中存在的讀取部分。

出于指導目的,我們再次通過以下方法簡化此問題:
僅考慮存在替換錯誤的區域,并忽略 indel 錯誤
僅考慮未存在已知變異的區域
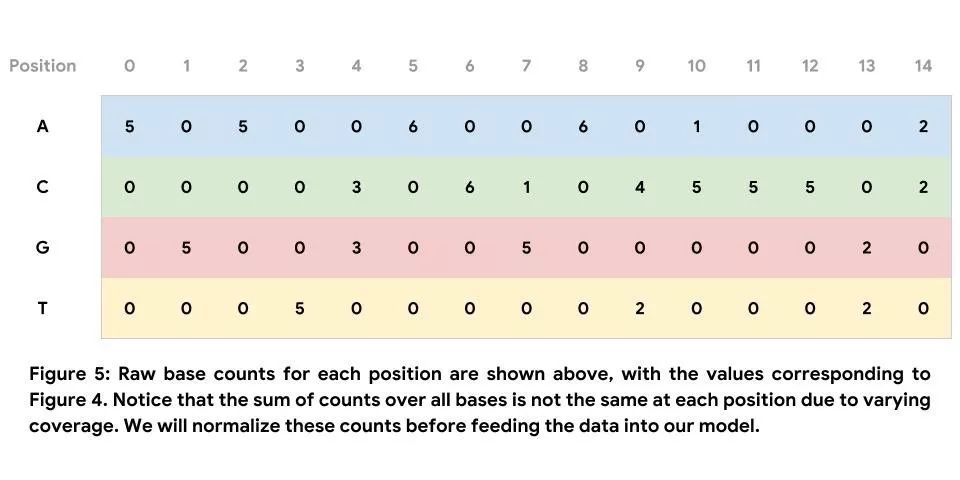
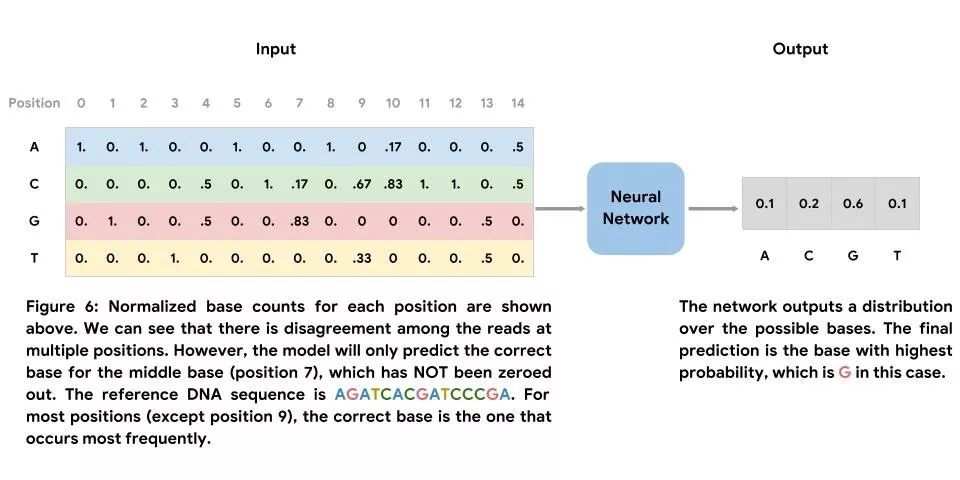
與第一種方法不同,我們并未在參考基因組中訓練此模型。相反,我們的訓練數據來自所映射的 Illumina HiSeq 讀取內容。此網絡的輸入是在所映射的讀取內容中觀測到的標準化堿基數矩陣,其核心是我們希望預測的正確堿基的位置。Clairvoyante(一種用于變異檢測的神經網絡)的作者以及Jason Chin 的示例方法中皆使用了類似的特征化方式。此網絡的輸出是可能出現的堿基分布,且最終預測結果為可能性最高的堿基。與第一種方法類似,我們使用在參考基因組中觀測到的堿基來產生標簽集。我們將包含錯誤(在堆疊中至少有一次讀取與中心位置中的參考序列不符)的示例及未包含錯誤(堆疊中的所有讀取均與中心位置中的參考序列相符)的示例結合使用。
結論
此隨附教程演示了本文所述的第二種方法。盡管我們分析的示例較為簡單,不適合在生產環境中部署,但我們希望它們能幫助開發者學會高效利用 Nucleus 和深度學習解決基因組學領域的問題。
-
機器學習
+關注
關注
66文章
8438瀏覽量
132930 -
深度學習
+關注
關注
73文章
5512瀏覽量
121415 -
tensorflow
+關注
關注
13文章
329瀏覽量
60593
原文標題:使用 Nucleus 與 TensorFlow 進行 DNA 測序糾錯
文章出處:【微信號:tensorflowers,微信公眾號:Tensorflowers】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
基于大數據的食品組學精進安全食品
全基因組數據CNV分析簡介 精選資料分享
全基因組測序的優勢 精選資料分享
使用Arm服務器減少基因組學的時間和成本
利用基因組學和人工智能延年益壽將在未來可期
AI加速推動醫療個體化轉型 基因組學將有望成為未來發展主流
為什么基因組是決定一個人特征的獨特遺傳密碼
基于計算分析的高可用彈性宏基因組學計算平臺
北鯤云超算平臺對于基因組學研究能夠提供哪些幫助?
NVIDIA Clara Parabricks助力基因組學和藥物研究
微流控芯片技術在單細胞基因組學研究中的應用






 工商網監
工商網監
評論