 紅磷負載Au單原子實現CO2光還原為C2H6
紅磷負載Au單原子實現CO2光還原為C2H6
研究背景
光催化CO2轉化因其在高附加值化學品生產中的潛在應用而受到廣泛關注。在光催化CO2還原中,生產雙碳 (C2) 產品非常困難,C-C鍵的形成需要克服比C-H鍵和C-O鍵形成更大的反應能壘。在沒有犧牲劑的條件下,通過CO2還原生產氣態多碳烴類產物(如C2H6)的報道很少。
具有獨特電子結構和豐富活性位點的單原子光催化劑引起了大量的關注。金屬單原子活性位點的設計和構建可以加速光生電荷的轉移和反應中間體的偶聯。然而,對于以金屬單原子為活性位點的催化劑,一個C1中間體需要脫附并遷移到另一個C1中間體并進行C-C偶聯,這不可避免地降低了C2的生成速率和選擇性。
因此,如果活性位點也涉及單個金屬原子附近的載體,將會更加有利于反應的進行。因此,還需要考慮CO2還原中間體與載體之間的相互作用。載體的類型也極大地影響了C-C鍵的耦合。具有豐富活性位點的合適載體可以有效縮短光生電子和C1中間體的遷移距離,促進C2產物的生成。
然而,迄今為止,常見的半導體催化劑載體由多種元素組成。這些元素作為單原子配位元素,使得單原子位點的配位環境多樣化且難以控制。相比而言,結構確定的單元素載體在催化機理研究中具有巨大的優勢。
紅磷(RP)是一種磷單質光催化劑,由于其制備簡單、穩定性高等優點,在光催化領域具有廣闊的應用潛力。RP具有下列優點: (i) RP的組成元素單一,可以為金屬單原子催化劑提供統一的配位環境。
(ii)與常見的單原子配位元素(N、O、S等)相比,磷(P)的電負性較低,因此可以通過P元素的配位來控制金屬單原子的電子密度。(iii)富電子的P原子可以有效地吸附CO2,并通過路易斯酸堿相互作用,與CO2中的O相互作用,促進C-O鍵斷裂,產生C1中間體。因此,設計構建RP負載的金屬單原子催化劑,以促進C1中間體的電荷分離和偶聯,是實現高活性和高選擇性地制備C2產物的潛在途徑之一。
成果簡介
清華大學李亞棟院士、王定勝副教授和中山大學胡卓鋒副教授(共同通訊作者)通過在紅磷 (Au1/RP)上引入一個Au單原子來誘導C-C偶聯。P具有較低的電負性,可以更好的吸附CO2。Au單原子附近的富電子P原子可以作為吸附CO2并促進CO2裂解的活性位點。Au單原子可以有效降低C-C偶聯的能壘,促進C2H6的形成。
研究亮點
1.Au1/RP催化CO2光還原生成C2H6的選擇性和轉換頻率分別達到96%和7.39 h–1;
2.Au單原子不僅可以促進光生電荷的轉移,也可以誘導C1中間體偶聯形成C2產物。
圖文導讀
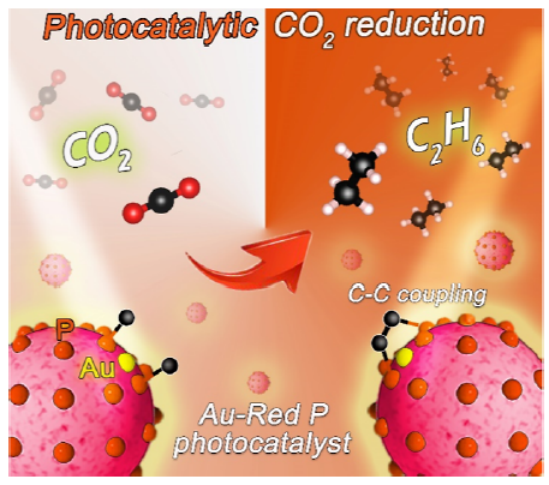
圖 1. Au 單原子與紅磷形成的界面輔助催化C-C偶聯。
將Au單原子負載到RP催化劑上,在沒有犧牲劑的情況下,光催化CO2還原為乙烷(圖1)。

圖 2. (a) Au1/RP的TEM。Au1/RP的AC HAADF-STEM (b) 和相應的放大圖像 (c) 。(d) Au1/RP的 HAADF-STEM 圖像。比例尺為100 nm。(e) Au1/RP的Au和P的元素映射圖。
如圖2a所示,Au1/RP的TEM表明,Au1/RP具有多孔形貌,未觀察到Au納米顆粒的存在。如圖2b、c所示,發現許多孤立的亮點,這些亮點被識別為Au 單原子。Au1/RP的HAADF-STEM顯示,沒有發現金屬顆粒(圖2d)。如圖2e所示, Au原子均勻分布在RP骨架上。
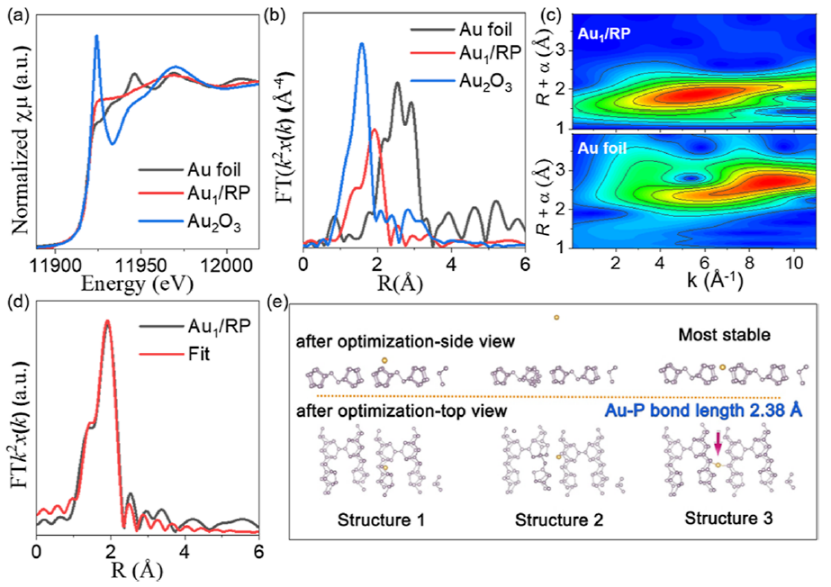
圖 3. (a) AuK邊XANES光譜;(b)傅里葉變換圖;(c)小波變換EXAFS 光譜;(d) Au1/RP在R空間的EXAFS擬合曲線;(e) Au1/RP三種可能的結構。
如圖3a所示, Au1/RP的XANES光譜吸收強度介于Au箔和Au2O3之間,表明Au1/RP催化劑中的Au物種帶正電,價態為在0和+3之間。從EXAFS分析(圖3b)可以看出,Au1/RP在1.83 ?處顯示出突出的峰。由于Au-Au 配位 (>2.5 ?) 的位置沒有觀察到峰,這表明Au1/RP 中不存在 Au 粒子。
從Au箔(圖3c)可以分析出 R 空間中2.7和1 ?處的最高強度峰對應于Au-Au和Au-O配位。然而,Au1/RP僅檢測到R空間中 1.8 ?處的峰值強度最大值(圖3c),這歸屬于Au-P配位。如圖 3d所示,Au1/RP的Au單原子配位數約為2。Au單原子與兩個P原子配位形成Au-P2結構體,平均鍵長約為 2.34 ?。
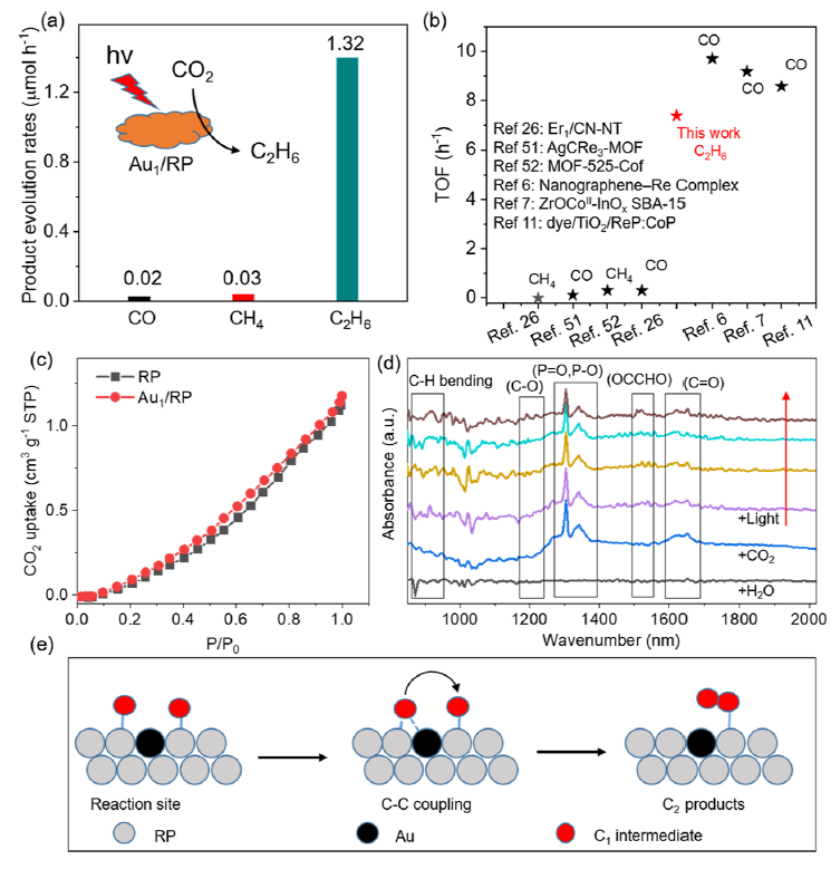
圖 4.(a) Au1/RP 在CO2還原反應中的光催化活性。(b)Au1/RP光催化CO2還原的TOF與近期報道的比較。(c) 室溫下,RP和Au1/RP的CO2吸附。(d)用于CO2還原反應的Au1/RP的原位DRIFTS光譜。(e) Au1/RP催化C-C偶聯的反應機理。
CO和CH4在Au1/RP上的產率分別僅為 0.02 和 0.03 μmol g–1h–1,而 C2H6的產率高達 1.32 μmol g–1h–1(圖 4a),選擇性高達 96%。Au/RP的 TOF可達到7.39 h–1(圖 4b)。如圖4c所示,負載Au單原子后,樣品對CO2的吸附能力沒有提高,證明Au不是CO2的吸附位點。如圖4d所示,在800–1700 cm–1處觀察到一些峰。1520 cm–1處的峰可歸因于OCCHO伸縮振動。900 cm–1處的峰與C–H的振動有關。
當添加CO2時,可以觀察到1357 和 1621 cm–1處的峰。這些峰分別對應于 P=O/P-O 和 C=O 的振動。這證明RP能有效吸附CO2分子,是CO2的活化位點。因此,可以推測Au1/RP催化C-C偶聯的反應機理。如圖 4e所示,Au單原子促進C1中間體在P反應位點發生解吸和遷移,然后在另一個P 位點進行 C-C 偶聯。
圖 5. 吸附了C-C中間體的Au1/RP的(a) 結構變化;(b) 能量變化。(c) 將CO2光催化還原為C2H6的三個重要步驟的能量變化。
光催化CO2還原反應過程中的能量變化如圖5a,b所示。在該反應的初始階段,反應體系的能量迅速下降,相鄰C原子間距變小(狀態1到狀態4)。從狀態4到狀態5,能量保持在同一水平。在此期間,形成了C-C鍵,兩個C原子也與附近的P原子成鍵。
隨后,一個C原子離開P原子并靠近Au原子(狀態 6 到狀態 8),這可能是由于Au對C原子的吸引。在此期間,能量再次下降。因此,Au1/RP界面有利于C-C原子偶聯及C2H6形成。作者發現,反應體系的能量從初始狀態 (-0.16 eV) 下降到穩定的中間狀態 (-1.26 eV),表明CO2偶聯很容易發生在 Au1/RP界面(圖 5c)。
然而,從中間態到最終態,體系能量增加,表明該反應需要克服能壘才能形成C2H6。因此,理論計算可以證實,單個金屬原子可以通過調控活性位點的電子結構,促進活性位點上C1中間體的解吸和遷移,進而與另一位點的C1中間體偶聯,導致較低的C-C偶聯能壘。
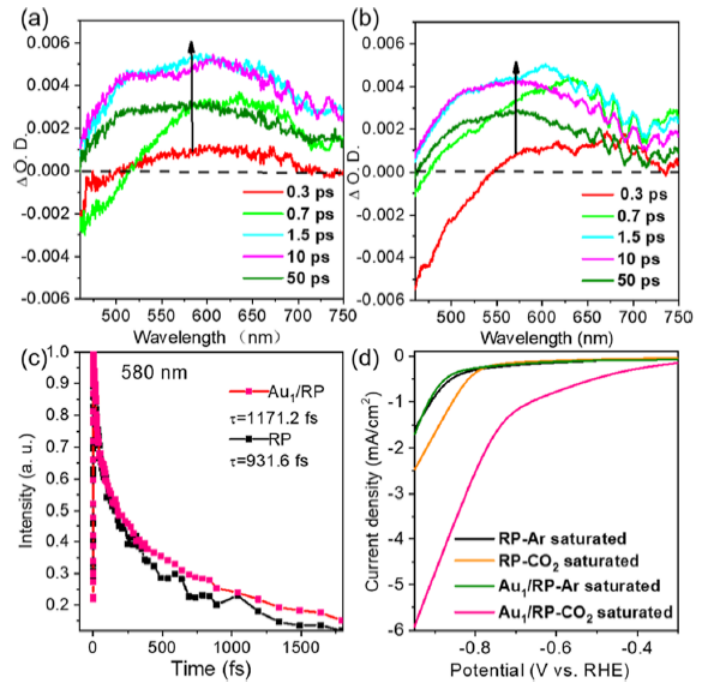
圖 6. (a) RP和 (b) Au1/RP的瞬態吸收光譜。(c)RP和 (b) Au1/RP在580 nm波長處的歸一化衰變曲線。(d) RP和Au1/RP在Ar或CO2飽和的KHCO3電解質中的線性掃描伏安曲線。
RP的瞬態吸收光譜如圖6a所示。在開始時(0.3ps),可以觀察到明顯的正吸收峰,這對應于發射態的光激發電子。隨著時間的推移(0.3-1.5 ps),這個峰大大增加,表明光生電子的積累。隨后,在50 ps時,由于空穴和電子的復合,觀察到輕微的衰減。
對于Au1/RP,正吸收峰強度的增加甚至更快(圖 6b)。在0.7ps時達到最高,一直保持到10ps。這表明Au單原子促進了光激發電子的產生。此外,光激發電子的壽命也可以通過 580 nm 波長下的歸一化單衰變曲線來確認(圖6c)。初始上升階段表示電荷載流子從基態到激發態是瞬時產生和直接激發的。
隨后的下降階段是由于電子和空穴的復合。Au1/RP比RP衰減得更慢,對應于更慢的電荷復合率。Au1/RP中的電子壽命為1171.2 fs,而RP中的電子壽命為931.6 fs。如圖6d所示, RP和Au1/RP有類似的曲線,由于飽和Ar中的陰極電流主要來自析氫反應,該結果表明Au不會促進析氫反應。相比之下,在CO2飽和的電解質中,Au1/RP的電流密度增加明顯更快,并且起始電位也更早。
總結與展望
作者用RP負載Au單原子,將CO2有效的光催化還原為C2H6。通過各種表征技術,包括AC HAADF-STEM、XAFS光譜和理論計算,分析了Au單原子的結構和化學性質。實驗和理論計算結果證實,RP上的Au原子不僅能有效促進C-C偶聯,還能促進光生電荷的轉移,從而促進CO2還原為C2H6。該研究為C1轉化提供了一種在原子水平上設計催化劑的活性位點,從而實現C-C偶聯的有效方法,對基于太陽能的燃料制備具有重要意義。
文獻鏈接
Ou Honghui et.al Atomically Dispersed Au-Assisted C–C Coupling on Red Phosphorus for CO2Photoreduction to C2H6(J. Am. Chem. Soc.2022, DOI:10.1021/jacs.2c09424)
文獻鏈接:
https://pubs.acs.org/doi/full/10.1021/jacs.2c09424
審核編輯:劉清
-
電解質
+關注
關注
6文章
820瀏覽量
20123 -
TEM
+關注
關注
0文章
89瀏覽量
10427 -
TOF
+關注
關注
9文章
485瀏覽量
36427
原文標題:李亞棟/王定勝/胡卓鋒JACS: 紅磷負載Au單原子實現CO2光還原為C2H6
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
紅外 CO2(二氧化碳) 氣體傳感器和分析模組

松下CO2焊機維修維修
貿澤開售可精確測量CO2水平的 英飛凌PASCO2V15 XENSIV PAS CO2 5V傳感器
新世聯科技:NG2-A-7在DAC空氣捕集提取CO2的應用
“不需要點表的工業網關”如何實現松下FPG-C32T2H數據采集和遠程維護的物聯網解決方案

利用單對以太網最大限度地減少 CO2 排放
英飛凌XENSIV PAS 5V CO2 傳感器概述

富昌電子推出英飛凌新品試用——基于光聲光譜 (PAS) 技術的創新性CO2傳感器
CO2 AI推出首個大規模產品排放量計算解決方案
紅外CO2傳感器ACD1100,精準監測,守護呼吸健康

控制型單節電池 5A 快速充電器I2C BQ25890H數據表

CO2激光相位延遲反射鏡參數介紹

2μm單縱模全固態脈沖激光技術研究進展綜述





 工商網監
工商網監
評論