 單原子Cu催化劑的重構現象以及它們在電催化反應的應用
單原子Cu催化劑的重構現象以及它們在電催化反應的應用
催化劑的重構現象普遍存在于許多多相反應。目前,借助先進的表征技術以及理論計算,我們已經能夠探索、理解多種催化劑的重構機制以及它們對催化反應的利弊。然而,對于單原子催化劑,其重構現象以及機理研究卻比較罕見。在許多情況下,我們都默認它們在多相催化反應中是穩定的,特別是在反應初始階段。
1. 電位驅動單原子Cu催化劑在硝酸鹽還原反應的結構演變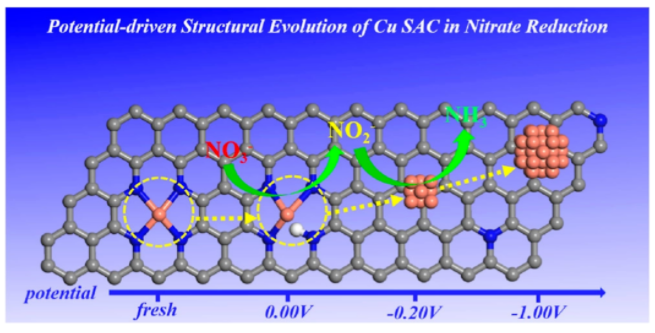
2.電位驅動單原子Cu催化劑在氧還原反應的結構演變
盡管單原子Cu催化劑的重構現象的研究取得了一定的進展,然而,由于催化反應不同,反應介質以及應用電位也存在差異,目前仍難以清晰解釋單原子Cu催化劑的重構現象。
最新成果介紹
德克薩斯大學奧斯汀分校劉遠越教授、東南大學王金蘭教授等人以單個Cu原子嵌入N摻雜石墨烯為例,利用“恒勢混合溶劑化動力學模型”,在實際反應條件下評估了單原子Cu與Cu團簇之間的可逆轉化。結果表明,H的吸附是單原子Cu從催化劑表面浸出的重要驅動力。電極電位越負,對H的吸附越強,競爭性析氫反應受到抑制,Cu-N鍵發生減弱,導致Cu原子部分被錨定在催化劑表面,部分溶解在水溶液中。
在兩種狀態下Cu原子發生碰撞、形成瞬時Cu團簇結構,成為促進CO2還原為乙醇的真正催化活性位點。當外加電位被除去或轉換為正電位時,羥基自由基(OH?)將進一步氧化Cu團簇,Cu通過再沉積、恢復到初始的原子分散狀態,最終完成催化劑的重構循環。
因此,該工作提供了對Cu單原子催化劑在工況下的動態穩定性的基本理解,并呼吁考慮現實的反應條件,重新評估目前報道的單原子催化劑的穩定性。
圖文介紹
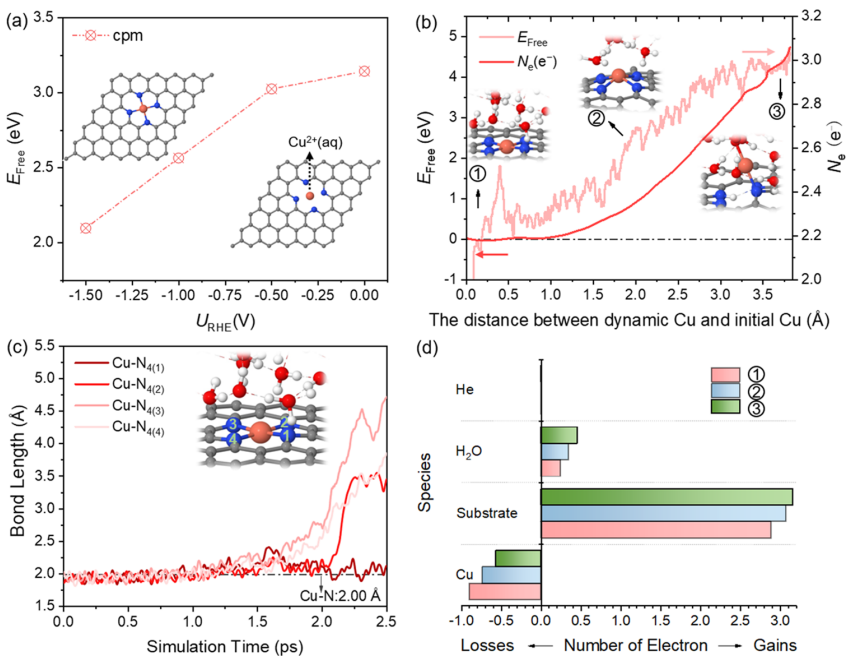
圖1. Cu浸出過程的熱力學和動力學分析
圖1a顯示了在0 ~-1.5 V的不同電極電位下,Cu SA從表面浸出、形成Cu2+(aq)的自由能。雖然在-1.5 V時自由能降低到2.09 eV,但從熱動力學上仍然很難從表面浸出。進一步通過恒勢混合溶劑化動力學模型評估了從表面浸出Cu的動力學可能性。如圖1b所示,隨著Cu遠離表面,自由能繼續增加,在反應結束時達到4.74 eV。這表明Cu-N鍵不易斷裂,Cu SA不能在室溫下從表面浸出。
圖1c跟蹤了Cu-N的鍵長隨Cu SA浸出過程的動態演變。注意,當Cu-N(3)鍵斷開時,靠近N(3)原子的一個水分子發生水解,反應結束時對應的構型為Cu原子只與一個N原子和一個?OH基團配位。圖1b顯示了電子數隨結構的變化而變化。與初始結構相比,由于OH -的形成,最終結構的凈電荷數增加到約0.7 e-。基于Bader電荷分析,如圖1d所示,Cu失去的電子數從初始結構的0.9 e-下降到最終結構的0.56 e-,說明Cu SA發生價態降低、在遠離表面處可被還原。因此,熱力學和動力學結果都表明Cu SA幾乎不可能直接從表面浸出。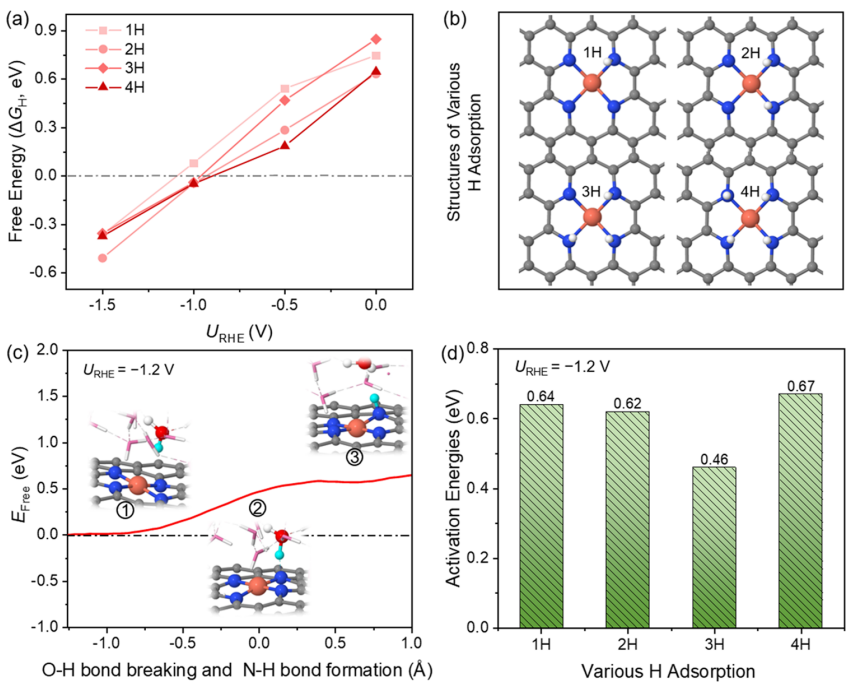
圖2.在催化劑表面的H吸附行為
然而,通過XAS表征,在原位CO2電解條件下原子分散的Cu2+和金屬Cu0團簇之間確實存在結構轉變。那么,具有強螯合能力的N4-C位點的Cu SA發生浸出、形成Cu團簇的驅動力是什么? 在此,探討了單原子Cu催化劑在CO2還原電位下H的吸附行為。圖2a、b分別顯示了不同外加電位下xH吸附的構型與自由能。
當電極電位由零變為負時,對應的H吸附自由能由正變為負,說明H在N位點的吸附從熱力學上由不利變為有利。這種轉變歸因于電位變得更負,導致更多的電子聚集在催化劑表面,從而促進H+的吸附。在U=-1.0 V時ΔGH接近于零,這解釋了為什么H2在-1.0 V時產率最高。在U=-1.2 V時,由于H吸附顯著增強,析氫反應(HER)被有效抑制。
在此,推測H的吸附可能是影響Cu在負電位下解吸的重要因素。 因此,進一步討論了H的來源。H的來源與H2O解離有關。因此,計算第一個H2O解離生成H*和OH-的動力學勢壘。在?1.2 V時能壘為0.64 eV,說明該反應在室溫下很容易發生。反應前后的電子數差約為0.8 e-,證實了OH-的生成。由于催化劑的結構中含有4個N原子,可以為H吸附提供4個活性位點,因此也計算了H占據的其余3個N位點的活化能,分別為0.62、0.46、0.67 eV。H2O解離生成*H和OH-的平均勢壘約為0.60 eV。因此,從熱力學和動力學兩個方面驗證了在U=-1.2 V時,H2O分子中的H可以被吸附到N位點上。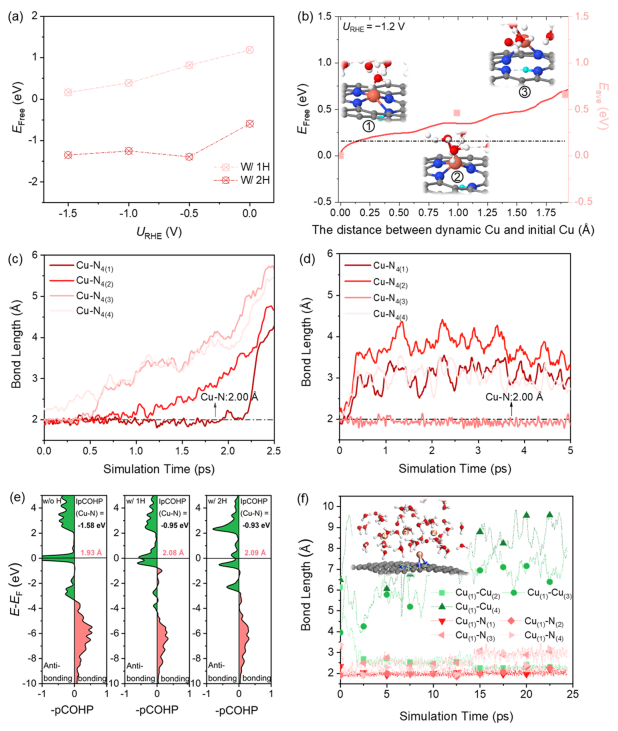
圖3. H吸附驅動Cu從催化劑表面發生浸出
接下來,需要考慮的是:在CO2還原過程中,H的吸附如何影響催化劑結構的轉變? 計算1H和2H吸附下Cu SA浸出過程中Cu2+(aq)形成的自由能,如圖3a所示。隨著H的吸附,Cu SA與基底的結合強度減弱,形成了有利于Cu SA浸出的熱力學過程。而對于動力學,重新評估了Cu-N4-C中Cu SA浸出過程的動力學勢壘,其中一個N位點被一個H位點吸附。如圖3b所示,當活化能為0.70 eV時,對應于Cu與一個N原子和一個H2O分子發生配位。與純Cu-N4-C表面(4.74 eV)相比,H的吸附顯著促進Cu SA的浸出。
如圖3c所示,當模擬時長為2.25 ps時,Cu原子完全脫離表面,溶解在水溶液中,并吸附兩個H2O分子。圖3d顯示了Cu-N4的鍵長(x,x=1~4)的動態演變,表明在2H共吸附條件下,Cu SA在短時間內(~ 300 fs)從表面自發浸出。所得到的最終構型由一個Cu-N鍵和至少一個Cu-O鍵組成。因此,在動態的電化學界面上,應該同時存在Cu與一個N原子結合的不完全浸出、以及溶解在水中的完全浸出的兩種瞬時狀態。 在動態環境下,兩種瞬時狀態的Cu原子發生碰撞形成瞬態Cu3/4團簇結構,成為真正的催化活性中心。進一步模擬了在工作條件下Cu原子的聚集過程。
從圖3f中可以觀察到,在AIMD過程中,兩個Cu原子的聚集小于2.5 ps, Cu(1)原子仍然錨定在N(1)和N(2)上。在15 ps時,Cu(1)-Cu(2)的鍵長進一步縮短為2.26 ?, Cu(1)-N(3)和Cu(1)-N(4)的鍵長分別拉伸為3.09和3.03 ?。一旦形成越來越多的Cu小團簇,它們可以加速CO2的還原、形成乙醇。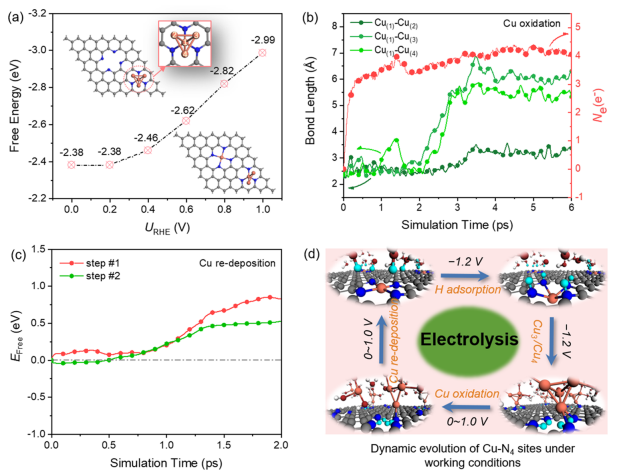
圖4. 在工況下Cu-N4位點的動態演化
CO2還原反應結束后,電極電位消失或升高至+1.0 V。此時,Cu團簇可以還原為原子分散的Cu2+。如圖4a所示,隨著電位的增加,Cu SA-Cu3生成的自由能越負,即Cu SA-Cu3的生成越有利。 最近,一項實驗研究證實,HCO3-與H2O之間發生快速氧交換,促進了高氧化性羥基自由基(OH?)的形成,從而促進了Cu的快速再氧化。
因此,考慮在體系中引入兩個OH?自由基來評估+1.0 V下Cu4團簇的再氧化過程。如圖4b所示,兩個OH?自由基分別在2 ps、0.5 ps后氧化Cu-Cu的第一配位殼層。在此之后,Cu(1)原子與其他三個或兩個Cu原子的距離越來越遠。因此,與純水溶液相比,OH?自由基的存在在Cu的快速再氧化中起著主導作用。在氧化過程中,系統的凈電荷處于相對平衡狀態,幾乎沒有額外的電子轉移到電極上,表明Cu團簇被OH?自由基氧化為Cuδ+。
當被OH?氧化后、形成Cu(H2O)3,此時Cu(H2O)3擴散到N4-C位點鄰接處時,即第1步,Cu從水溶液中遷移到與N原子配位,動能勢壘為0.85 eV,發生了Cu再沉積過程。在這個反應過程中,3個H2O分子返回水溶液中,Cu-N鍵的平均鍵長為1.94 ?,與初始Cu-N鍵長(1.95 ?)一致。也就是說,一旦Cu原子配位到1個N,Cu原子很快就會回到它最初的分散狀態。因此,在正電位下,高氧化的OH?與N4-C位點強螯合能力的協同作用,促進Cu團簇恢復到Cu SA狀態,完成循環。
審核編輯:劉清
-
電極
+關注
關注
5文章
821瀏覽量
27250 -
電荷
+關注
關注
1文章
631瀏覽量
36172 -
負電位
+關注
關注
0文章
4瀏覽量
5564
原文標題:JACS:單原子Cu催化劑在電催化反應中的結構演變
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
燃料電池的主要材料 燃料電池的效率和性能


理濤-催化劑磨損指數測定儀 催化劑磨耗測試儀-視頻解說 #催化劑磨損指數測定儀 #催化劑磨耗測試儀 #理濤
微反應器在有機合成及催化中的應用
實時原位監測光電催化過程中反應物濃度與熱效應的微光纖傳感器技術
中國科大在納米級空間分辨紅外成像研究中取得新進展
中國科大在納米級空間分辨紅外成像及催化研究中取得新進展
催化燃燒設備數據采集遠程監控系統解決方案
網關助力催化劑產業升級,解決痛點問題!

浪潮通信信息榮獲2024 TM Forum催化劑項目大獎
EthernetiP轉modbusTCP網關在加氫催化中的應用

相調控對鎳錫合金的電催化氮還原調控機制研究





 工商網監
工商網監
評論