

分子動力學(MD)模擬對于研究小分子和大型生物分子系統(tǒng)的構象移動性非常有用,但對于MD力場預測的構象能量和分布的準確性仍存在疑慮。有令人鼓舞的跡象表明,MD可以合理預測構象之間的轉變時間尺度,但這一成功必須得到驗證:除了少數(shù)例外,力場參數(shù)通常設定為重現(xiàn)熱力學性質和量子力學能量,而不是構象轉變的速率或停留時間。隨著開發(fā)出具有正確自擴散和粘度的新水模型,以及探索對蛋白質非結構區(qū)域建模的新方法,徹底的實驗驗證變得越來越重要。
一、寫在文前
磁共振在探測構象及其動態(tài)特性方面具有獨特的能力,尤其通過自旋弛豫和交叉弛豫測量。然而,大多數(shù)用于解釋自旋弛豫數(shù)據(jù)的模型在局部移動性方面做出了簡化的近似,并使用了孤立自旋對模型。最近出現(xiàn)了更為復雜的利用MD模擬進行NMR和EPR弛豫的方法,但它們仍然主要使用自旋對近似;我們小組的工作也屬于這個類別。需要的是一個軟件框架,能夠將MD軌跡直接轉換為任意自旋系統(tǒng)的弛豫超算符。
鑒于此,本文報告了在Spinach庫中實現(xiàn)了這個功能。我們在使用OPC和TIP5P水的甘露糖(GLYCAM06力場)上測試了所提出的方法。甘露糖的構象動力學已經有了較好的研究,尤其是通過使用孤立自旋對近似解釋的13C自旋弛豫測量。
在這里,我們將重點放在1H-1H核Overhauser效應(NOE)上,這種效應探測葡萄糖的H1環(huán)異構體質子(圖1)和連接葡萄糖和果糖殘基的糖苷鍵兩側的果糖H11,12質子之間的接觸。我們證明,從1微秒長的MD軌跡中提取的(交叉)弛豫率與實驗數(shù)據(jù)非常吻合,考慮到NOE差異為9-25%,這會轉化為1.5-4.2%的距離誤差。這對于GLYCAM06力場以及OPC和TIP5P水模型來說是一個好消息。核自旋(交叉)弛豫率很容易測量,我們建議將這樣的測試納入新型分子動力學力場的標準基準測試集中。相關工作以“Using molecular dynamics trajectories to predict nuclear spin relaxation behaviour in large spin systems”發(fā)表在《Journal of Magnetic Resonance》上。
二、圖文速遞
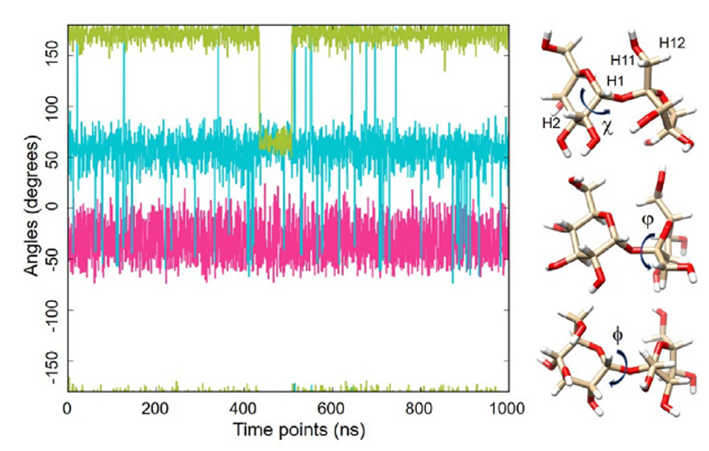
圖1. 甘露糖內所示二面角的動力學特性(1毫秒MD軌跡,GLYCAM06甘露糖,OPC水,300K)。v(H2-C2-C1-O20)以綠色顯示,u(C1-O20-C2-C1)以青色顯示,/(H1-C1-O20-C2)以紅色顯示。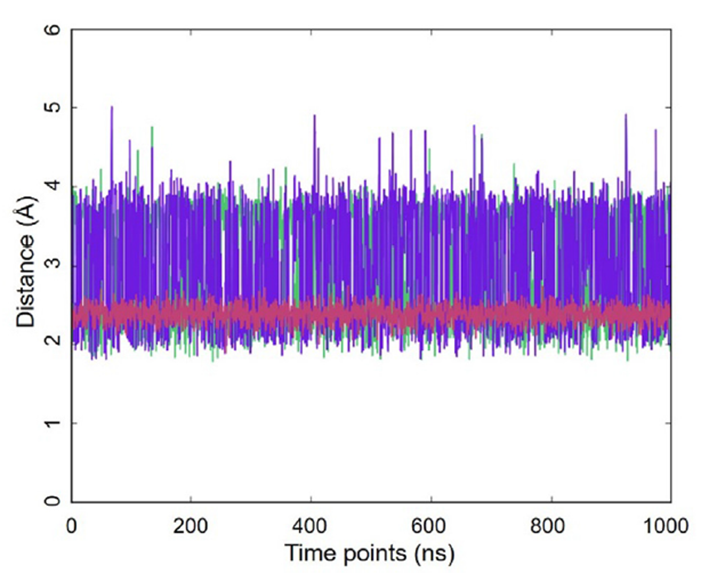 ?
?
圖2. 原子之間的距離動態(tài)(1毫秒MD軌跡,GLYCAM06甘露糖,OPC水,300K):葡萄糖H1到果糖H11的距離(綠色),葡萄糖H1到果糖H12的距離(紫色),葡萄糖H1到葡萄糖H2的距離(紅色)。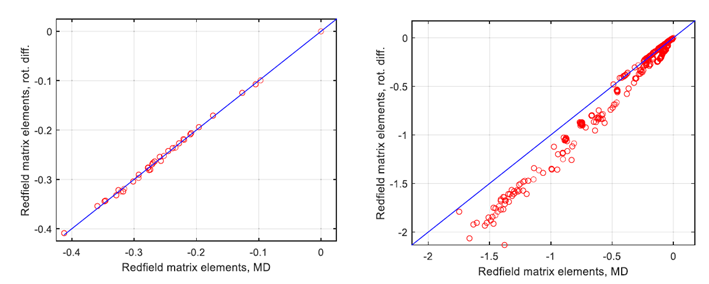
圖3. 對比分析處理(使用純旋轉擴散近似)和數(shù)值處理(使用MD軌跡積分,OPC水)中弛豫超算符的非零元素之間的差異,隨著內部運動對自旋弛豫的貢獻增加。左側面板:相對剛性的葡萄糖環(huán)中的3個自旋子系統(tǒng)(H1,H2,H4),其中偶極耦合調制幾乎完全是旋轉性質。(右側面板)包含6個自旋子系統(tǒng)(葡萄糖的H1,H2,H3,H5;果糖的H11,H12),其中存在與糖苷鍵和外環(huán)C1-C2果糖鍵相關的顯著內部運動。兩個面板中的藍線對應旋轉相關時間為78皮秒,該值是通過擬合左側面板的數(shù)據(jù)獲得的。
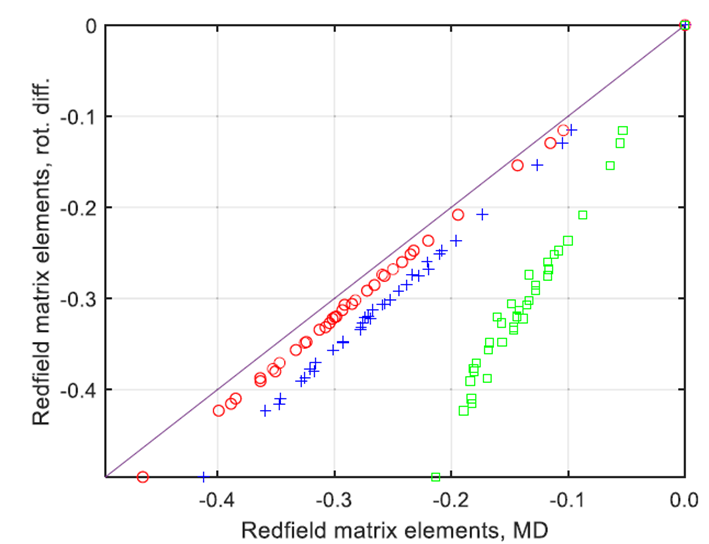
圖4.來自TIP3P水(綠色方塊)、TIP5P水(紅色圓圈)和OPC水(藍色十字)的分子動力學軌跡所得到的Redfield弛豫超算符的非零元素(X軸),與使用純旋轉擴散近似和旋轉相關時間為100皮秒獲得的相應元素的解析弛豫超算符進行對比(綠色方塊代表TIP3P,紅色圓圈代表TIP5P,藍色十字代表OPC)。
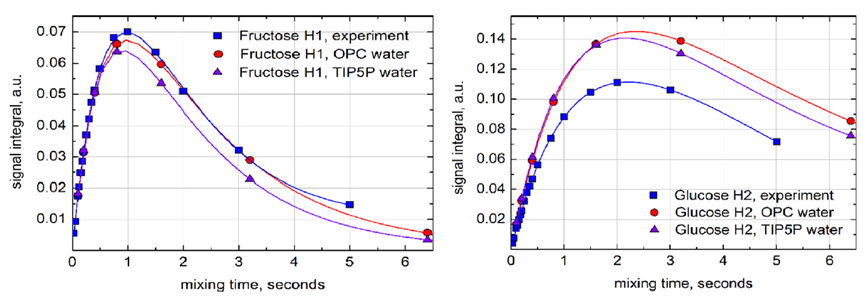
圖5. 在葡萄糖H1質子自旋反轉后,實驗(藍色)和模擬(OPC水為紅色,TIP5P水為紫色)的核Overhauser效應(NOE)累積曲線的比較(曲線為雙指數(shù)擬合)。模擬使用以葡萄糖H1為中心的8個自旋系統(tǒng)進行,包括(Glc H1-H5;Fruc H11,H12,H3)。信號積分是反轉共振強度的分數(shù)。實驗數(shù)據(jù)在600 MHz和308 K下收集;選擇更高的溫度是為了補償D2O的增加粘度。沒有嘗試補償蔗糖濃度對粘度的影響-預計該影響在0.5%以下。在進行核磁共振實驗之前,樣品被除氣處理。
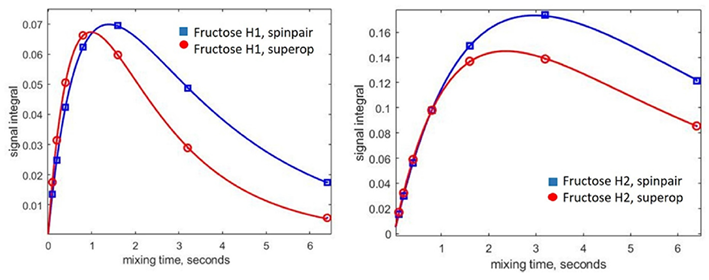
圖6. 使用自旋對(藍色)和完整自旋系統(tǒng)(紅色)方法對模擬的核Overhauser效應(NOE)累積曲線進行比較(在葡萄糖H1共振反轉時)。實線是雙指數(shù)擬合。左側面板:果糖H11 + H12質子。右側面板:葡萄糖H2質子。
三、小結
我們提出了一個新工具,用于將實驗自旋弛豫數(shù)據(jù)與基于分子動力學軌跡的模擬進行比較。它通過對Redfield積分進行數(shù)值評估直接計算自旋弛豫超算符。該工具已集成到Spinach軟件包中,并能夠模擬多種自旋弛豫實驗,包括具有顯著強耦合和弛豫干涉效應的自旋系統(tǒng)。這類計算直到最近才變得可行,其中有五個主要因素起到了貢獻: (a)具備足夠處理能力的圖形卡的易得性(NVidia Ampere架構超過10TFLOPS的處理能力,約等于2002年全球最大超級計算機); (b)在磁共振中開發(fā)和實施了用于弛豫超算符和稀疏矩陣指數(shù)的大規(guī)模方法; (c)Matlab的性能和語法靈活性不斷提高,其中的程序代碼通常比用人類語言和標準數(shù)學符號描述更短且更易讀; (d)出現(xiàn)了線性復雜度縮放的液態(tài)自旋動力學模擬方法,使得蛋白質大小的系統(tǒng)不再無法達到; (e)提供了足夠長的分子動力學軌跡,以同時在方程中對上限和集平均值進行收斂。
我們通過在蔗糖上模擬NOE實驗對該工具進行了測試,使用了GLYCAM06力場下的1毫秒分子動力學軌跡和OPC、TIP5P和TIP3P水模型。與實驗的一致性非常好,考慮到0-25%的NOE初始斜率的差異對應剛性結構中的距離誤差為0-4.2%。這對于GLYCAM06糖類參數(shù)化和OPC、TIP5P水模型來說是非常好的,但TIP3P效果不佳。上述模擬的CPU時間需求不超過運行分子動力學軌跡所需的時間。一旦計算了弛豫超算符,通過計算多個混合時間下的NOE或模擬任何其他弛豫實驗所消耗的CPU時間是微不足道的。在具有24個CPU核心的系統(tǒng)中,使用IK-0(3)基組和每2皮秒采樣一次的1皮秒軌跡,8自旋弛豫超算符計算大約需要30小時;如果使用GPU,這個時間將大大縮短。預計更大的自旋系統(tǒng)將變得簡單:一旦自旋數(shù)超過受限態(tài)空間近似中的強相互作用團簇的大小(通常在液態(tài)NMR模擬中少于五個自旋),計算復雜性與自旋系統(tǒng)的大小大約呈線性關系。
審核編輯:劉清
-
gpu
+關注
關注
28文章
4880瀏覽量
130342 -
OPC
+關注
關注
7文章
355瀏覽量
46855 -
NMR
+關注
關注
0文章
10瀏覽量
7003
原文標題:Andy博士:使用分子動力學軌跡來預測大型自旋系統(tǒng)中的核自旋弛豫行為
文章出處:【微信號:sim_ol,微信公眾號:模擬在線】歡迎添加關注!文章轉載請注明出處。
發(fā)布評論請先 登錄
相關推薦
Adams多體動力學仿真解決方案全面解析
電磁軌跡預測分析系統(tǒng)軟件全面解析
輪轂電機驅動電動汽車垂向動力學控制研究綜述
航空發(fā)動機整機動力學有限元模型建立方法
Nat. Mater.:室溫下PdSe?誘導的石墨烯平面內各向異性自旋動力學

致真精密儀器自旋測試多功能克爾顯微鏡介紹

弛豫頻率與截止頻率計算

【Simcenter STAR-CCM+】通過快速準確的CFD仿真加速空氣動力學創(chuàng)新

使用Phase Lab鎳基動力學數(shù)據(jù)庫計算多組分合金的成分分布曲線
自旋極化:開創(chuàng)半導體器件設計的新路徑





 工商網監(jiān)
工商網監(jiān)

評論